 infobox@neuroproof.com
infobox@neuroproof.com +49 381 54345-660
+49 381 54345-660
SCN1A – Dravet Syndrome Assay
4-AP model Developmental model
Loss of function of the SCN1A gene is the cause of Dravet Syndrome a catastrophic form of Epilepsy or other forms of Epilepsy and other diseases as such genetic epilepsy febrile seizures plus (GEFS+), Doose syndrome, West syndrome, Lennot-Gastraut syndrome, Rett syndrome, and nonsyndromic epileptic encephalopathy, as well as non epileptic diseases such as hemiplegia migraine and autism spectrum disorder (ASD).
There are existing more than 1000 SCN1A mutations with loss or gain of function. A knock down of the SCN1A would allow a phenotypic screening against loss of function.
SCN1A gene loss impairs voltage gated sodium channels NaV1.1 mainly on GABA-ergic neurons.
Example:
Cell Culture: Experiments were performed with primary frontal cortex cultures of mouse. SCN1A knock down were performed with an AAV shRNA construct.
We offer this assay also with human iPSC-derived neuronal cell cultures.
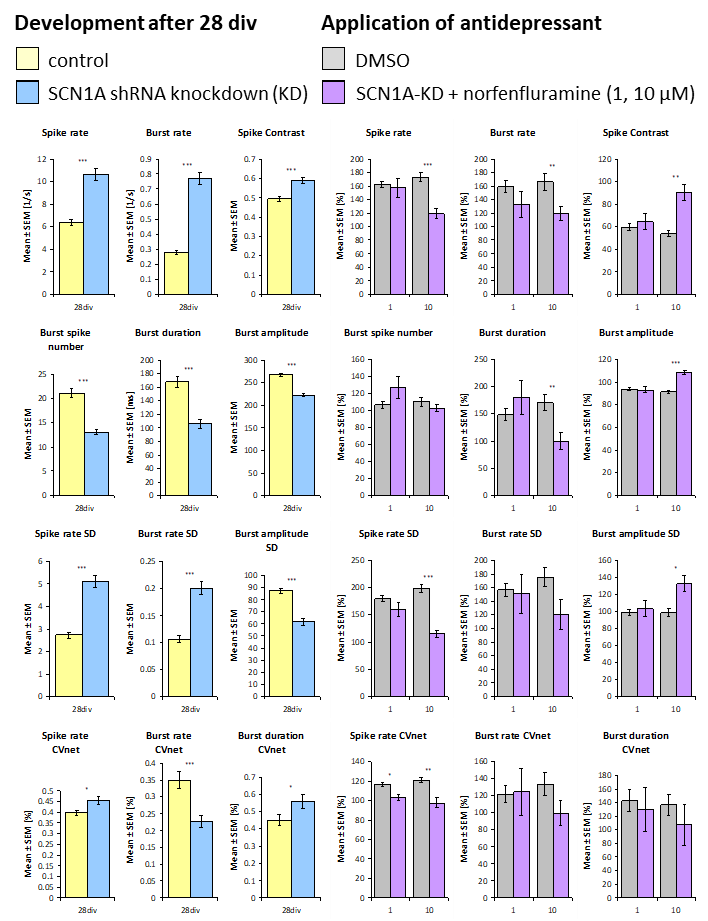
Image: Multi-parametric characterization of neuronal activity after norfenfluramine [µM] treatment. Mean values presented with standard error. Student’s unpaired t-test (* p<0.005).
Explore the comprehensive suite of assays to delve deeper into neurodegenerative mechanisms and potential therapeutic interventions..